
Article by Xin Li lab
Like animals, plants are constantly exposed to bacteria, viruses, and fungi that have the potential to cause diseases. To remain healthy plants possess a complex immune system which relies on immune receptors that are able to recognize pathogens and respond before an infection is established. If a pathogen is able to avoid detection by the immune system, the resulting plant disease can result in dramatic economic losses. An estimated 15% of potential crop output is lost to plant diseases, and periodic disease outbreaks can lead to large-scale social turmoil, such as the Irish potato famine1. Despite the importance of the plant immune system, the molecular details of immune signalling are only partially understood. Thus, the study of plant-microbe interactions is an active area of research, often involving the model plant Arabidopsis thaliana.
Plants have cell-surface receptors to detect common molecules produced by microbes, such as components of bacterial flagella and fungal cell wall polymers. However, many plant pathogens produce effector proteins that hamper this recognition. In response to this, higher plants have evolved Resistance (R) proteins that act as sensors to detect specific effectors. They may bind to effectors and recognize them directly, or they may recognize the changes that the effectors cause on other host proteins. Recognition of the effectors leads to activation of the R proteins, culminating in a defense response that effectively prevents the spread of the pathogen into healthy tissue.
Although plant cells must contain high enough levels of R proteins to quickly recognize invading pathogens, plants must carefully regulate the levels of these proteins in order to optimize growth, because excessive R protein accumulation may activate the defense responses even in the absence of pathogens. This is detrimental to the plant, as exemplified by the snc1 mutant of Arabidopsis identified by UBC professor Dr. Xin Li2,3. The snc1 mutant produces a form of an R protein that is more resistant to protein degradation, and therefore accumulates to higher levels in the plant.
In an attempt to search for genes which are involved in immunity, a forward genetic screen was performed looking for mutants which enhanced the autoimmune phenotype of snc1; this was named the muse (mutant, snc1-enhancing) screen4. From this screen a variety of mutants were identified, many of which play roles in R protein turnover. One of the mutants, muse6, was identified to be a subunit of an N-terminal acetyltransferase (Nat) complex5. N-terminal modification has been a long documented method of proteins regulation in plants as well as other eukaryotic organisms6.
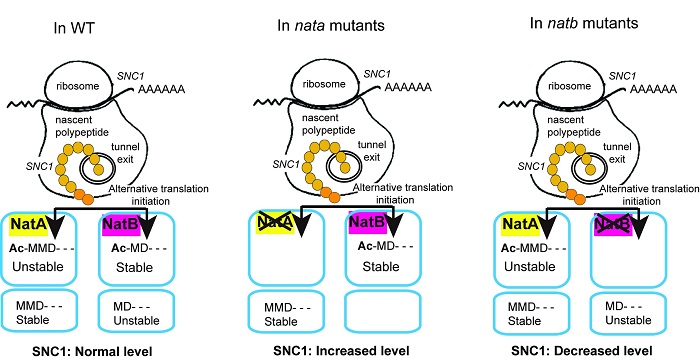
Two recent UBC Botany graduates, Dr. Fang Xu and Dr. Yan Huang, made most of the findings in the story. This discovery led to a detailed study of the N-terminal modifications of SNC1. What was discovered is that SNC1 has a rather unusual pattern of N-terminal modification. The SNC1 protein was found to have alternative initiation, meaning that its translation can begin from either of two adjacent methionines (Met); interestingly, SNC1 appears to be differentially regulated depending on which Met is used as the start codon. If SNC1 translation begins at the first Met it is acetylated by NatA, the Nat complex of which MUSE6 is a component, whereas if it begins at the second MET it is acetylated by a different complex, NatB.
The result of this alternative acetylation has unexpected consequences on SNC1 regulation. The initial screen demonstrated that disrupting NatA caused an increased autoimmune phenotype, and this is because acetylation of the first Met of SNC1 targets the protein for degradation. Alternatively, the acetylation of the second Met by NatB was found to stabilize SNC1. This interesting combination of alterative initiation and differential acetylation has never been reported until now. This novel pattern of alternative acetylation is likely one factor allowing for careful regulation of SNC1 protein levels, and by acting at the level of translation it allows for rapid changes in protein levels during the induction of defense responses.
1. Dangl, J. L., Horvath, D. M. & Staskawicz, B. J. Science 341, 746–751 (2013).
2. Li, X., Clarke, J. D., Zhang, Y. & Dong, X. Mol Plant Microbe Interact 14, 1131–9 (2001).
3. Zhang, Y., Goritschnig, S., Dong, X. & Li, X. Plant Cell 15, 2636–2646 (2003).
4. Huang, Y. et al. Nat. Commun. 4, 2558 (2013).
5. Xu, F. et al. Plant Cell. May 12. pii: tpc.15.00173 (2015). [Epub ahead of print]
6. Gibbs, D. J., Bacardit, J., Bachmair, A. & Holdsworth, M. J. Trends Cell Biol. 24, 603–611 (2014).
Figure 1. A nata mutant, muse6-1, identified from a genetic screen to search for enhancers of snc1.
Figure 2. A working model on the different N-terminal acetylations of SNC1. In wild type plants, SNC1 protein synthesis can be initiated at either the first or the second Met. The peptide translated from the first Met can be acetylated by NatA, which serves as a degradation signal. The peptide translated from the second Met can be acetylated by NatB, where the protein is stabilized by the modification. SNC1 protein homeostasis is maintained by the coexistence of these different protein isoforms. In nata mutants including naa15-1, aminaa15 and aminaa10 plants, almost all the SNC1 peptides are acetylated at the second Met by NatB due to the inefficiency of NatA. The stabilization by acetylation on the second Met leads to increased SNC1 accumulation, partly leading to the autoimmune phenotypes of these plants. In natb plants, as a result of inefficient acetylation of the second Met of SNC1 by NatB that stabilizes the protein, SNC1 protein levels are decreased.